How do piezo motors work?
Reading time: 8 min
There is a clear trend that piezo motors are being used more and more, as an alternative for electro motors, but also as an enabling technology for new applications. This is because R&D staff are getting more familiar with the technology and its advantages. But how do piezo motors work? We will explain it in this article.
Recently, Youtuber 'Know Art' visited Xeryon in his search to better understand precision mechanics.
The result? A deep dive into high-precision motion, and a great breakdown of our revolutionary technology:
Generally speaking, there are three types of piezoelectric motors, all of which are based on piezoceramic (PZT ceramic). If you want to know more about this material, please see this link. The most common type is the impact-driven stick-slip piezo motor. A second category consists of the stepper type of piezo motors, also called walking piezo motor, which are typically used for high-force applications.
The third type is the ultrasonic or resonant piezo motor. All three types have their specific advantages and uses, which can be explained by examining their working principle in more detail.
Stick-slip piezo motor (= inertia piezo)
Figure 1 explains the working principle of the stick-slip piezo motor. The upper sketch, marked with ‘1’, shows the different components of a typical stick-slip piezo motor stage. It consists of a piezo stack which is fixed on one side, a contact point, a slider (i.e. the moving part) and a bearing.
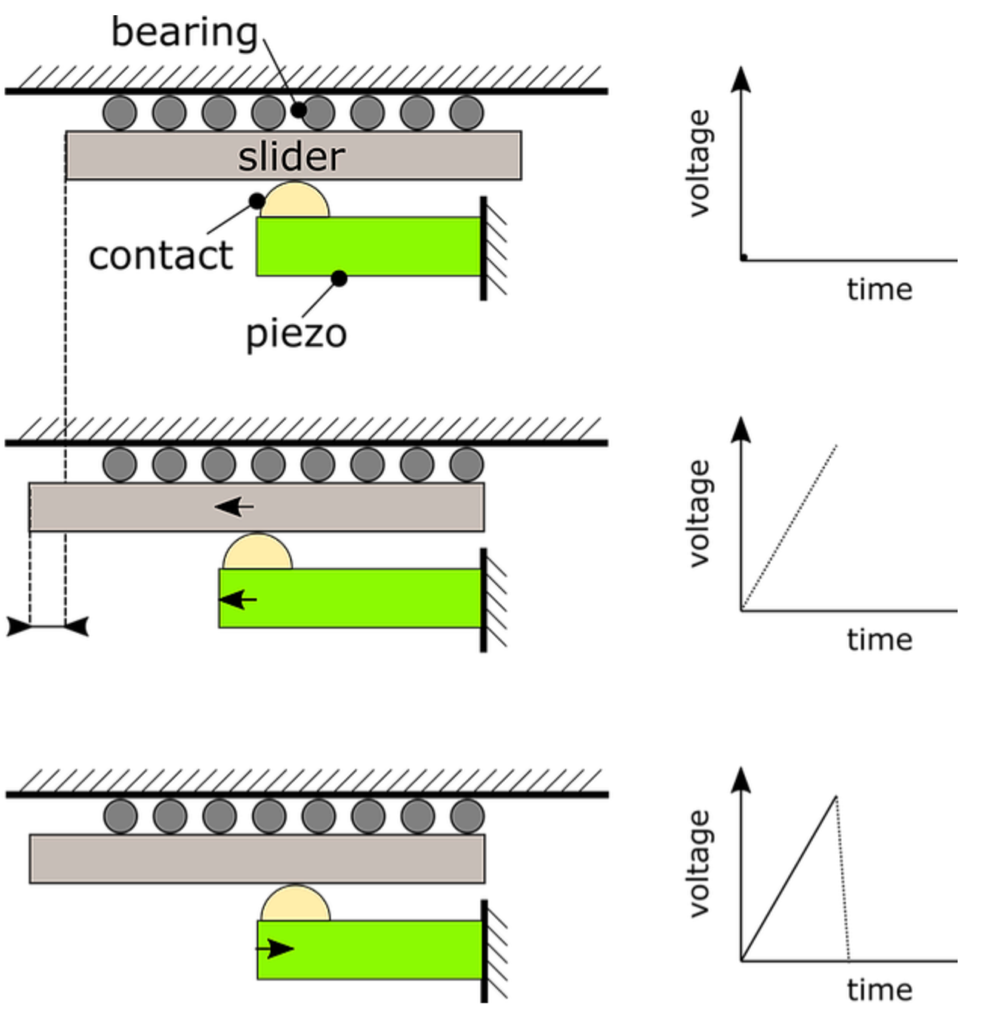
Figure 1: Working principle of a stick-slip piezo motor.
During the ‘stick-phase’, which is shown in state 2, the piezo is slowly extended because of a slow increase in voltage. The slider moves together with the moving contact point because of the friction force between the contact point and slider. Then, the piezo actuator is rapidly retracted by applying a rapidly decreasing voltage, see state 3 in Figure 1. Because of the inertia of the slider, it remains stationary and the contact point slips back to its original position. This is called the ‘slip-phase’ and results in a net displacement of the slider. By repeating these two steps, a macroscopic movement is realized.
A very similar type of piezoelectric motor is called the ‘inertial’ piezo motor. Although the driving signals are identical to the stick-slip principle, the inertial motor does not have a slipping contact point. More information about this type of motor can be found in “Piezoelectric Inertia Motors – A Critical Review of History, Concepts, Design, Applications, and Perspectives” (Hunstig, 2017).
Characteristics of stick-slip piezo motors:
The impact on the stage, which occurs during the slipping phase, causes an excitation of the system dynamics. This generates vibrations and noise. The noise can be very irritating and sometimes even cause health issues, especially when humans need to operate in the vicinity of the device. The impact-based driving mechanism also causes a significant amount of wear in the contact materials, typically limiting the lifetime of this type of stages. A stick-slip motor is characterized by a minimum step size during motion and it is virtually impossible to achieve a high repeatability because the step size depends on many operating conditions (for instance the motion direction). Some stick-slip motors use a DC scanning mode to realize a very fine resolution. Although this is effective to perform fine positioning, it becomes impossible to keep the final position stable at nanometer level with zero drift, or without powering the motor. The speed of most common stick-slip piezo motors is limited to about 20 mm/s. Researchers from the Paderborn University in Germany have written a review paper, which presents a more detailed overview of all commercially available stick-slip and inertial piezo motors. (Hunstig, 2017)
Stepping piezo motor (= walking piezo or piezo legs)
The basic working principle of the stepping piezo motor is explained in Figure 2.
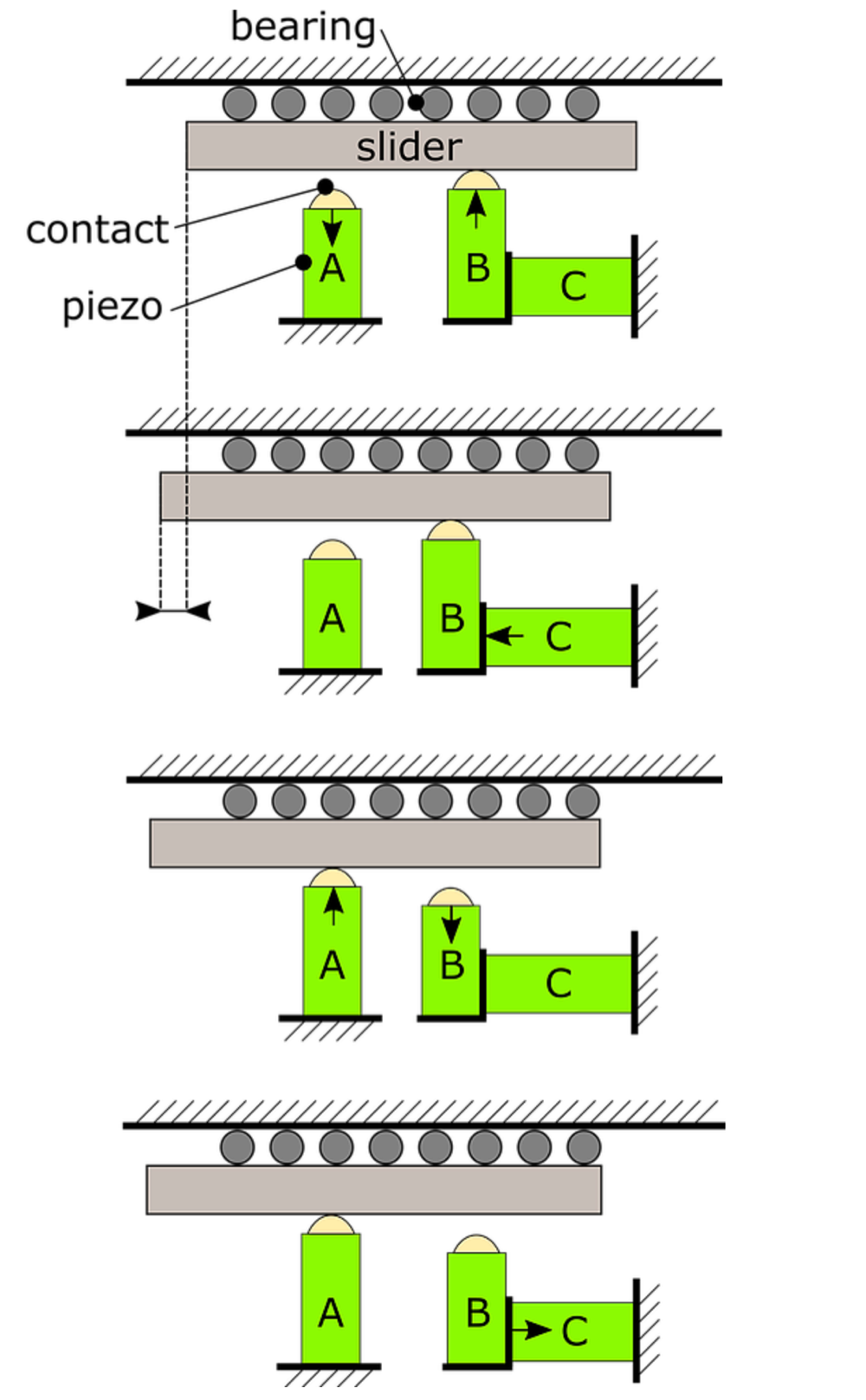
Figure 2: Working principle of a stepping piezo motor.
A typical stepping piezo motor consists of at least three piezo actuators. Some of the actuators are in contact with the slider (i.e. actuator A and B in Figure 2) and act as a clamping mechanism, while others are used to generate the translational motion of the slider (i.e. piezo C in Figure 2).
At rest, both piezos A and B are in contact with the slider. During the start of a motion cycle, which is shown in state 1 in Figure 2, piezo A is retracted while piezo B is expanded. Only the contact point of piezo B remains in contact with the slider.
Piezo C is expanded to realize the translational motion of the slider via the contact of piezo B, which is shown in state 2.
Next, piezo A is extended and piezo B is retracted. Now only piezo A is in contact with the slider, see state 3.
Subsequently, piezo C is retracted to its original position, resulting in state number 4.
Finally, piezo B is extended again and piezo A is retracted, restoring the initial conditions of the first state.
Characteristics of walking piezo motors:
Because of the different steps involved in the moving mechanism, a piezo stepper motor typically achieves low travelling speeds (i.e. < 10 mm/s). Next to that, during stepping, the contacts will wear off like in any kind of frictional contact. The piezo stepper motor however is particularly prone to wear, because a good operation requires strict tolerances. The reason for this is the small stroke of the piezo actuators, which need to be aligned carefully to each other. Therefore, piezo stepper motors typically achieve lifetimes smaller than for the other types of piezoelectric motors. A well-known and favourable configuration of a piezo stepper has four ‘legs’, but in total this motor consists of 8 different piezo actuators. Because of the large amount of piezos and the strict tolerances needed, this type of motor is more expensive than stick-slip and ultrasonic piezo motors.
Ultrasonic resonant piezo motor (with standing wave)
With this type of piezo motor, the motion of the slider is generated by an elliptical oscillation of the contact point(s). There are two types of ultrasonic piezo motors: the standing wave and travelling wave ultrasonic piezo motor. In a travelling wave motor, the contact point(s) shift(s) along the motion direction, constantly pushing the slider forward. The standing wave motor, on the contrary, has one (or a few) defined contact point(s). These contact points vibrate in an elliptical trajectory against the slider, resulting in a net displacement. Although the travelling wave piezo motors achieve high forces, they are prone to wear and have a limited lifetime. Therefore, this article focuses on the standing wave type piezo motor, which is the core competence of Xeryon.
In case of the standing wave type, the motion of the contact point is enhanced because the oscillation frequency coincides with two resonance frequencies of the motor structure. Figure 3 shows an example of the resonance modes of an ultrasonic piezo motor, which was developed by Xeryon’s founders at the university of Leuven in Belgium [2]. In the first resonance mode, the contact point moves in the tangential direction, i.e. the direction of motion. This mode is sometimes referred to as the ‘bending mode’. In the second resonance mode, called the ‘normal mode’, the contact vibrates perpendicularly to the direction of motion.
Figure 3: Resonance modes of an ultrasonic standing wave piezo motor
Animations: Know Art

Figure 4: Ultrasonic motor driving our XLA micro linear actuator
By carefully selecting the electrical drive signals of the piezo motor, both modes are excited simultaneously, with a phase difference of plus or minus 90 degrees. This leads to an elliptical vibration of the contact point, as shown in Figure 4. Next to the phase difference, the motion trajectory of the contact point can also be controlled by the amplitude or frequency of the drive signals.
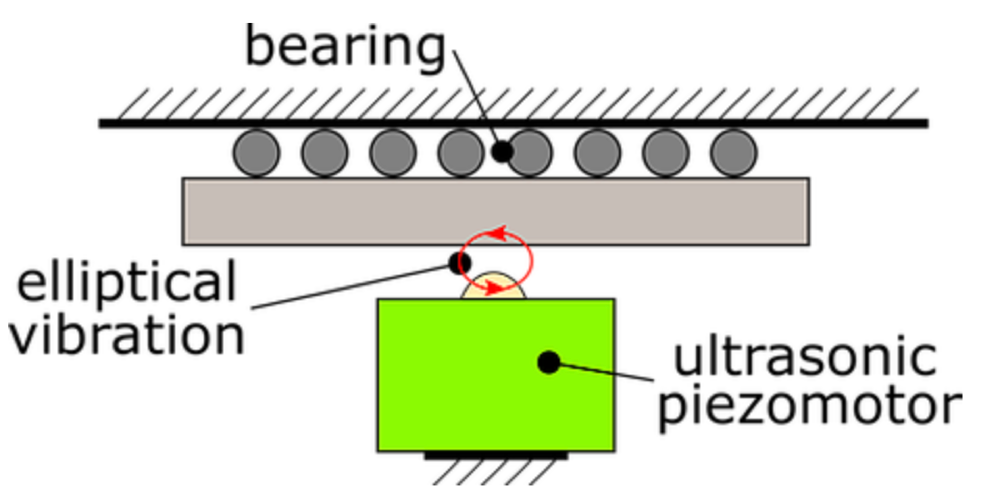
Figure 4: Elliptical vibration of the contact point of an ultrasonic piezo motor.
Characteristics of ultrasonic piezo motors:
The elliptical trajectory of the contact point can be shaped in such a way, as to have a very small horizontal amplitude, resulting in a small horizontal contribution of motion. This leads to a very fine positioning resolution, without the need for an additional quasi-static scanning mode (which is typically used in stick-slip piezo motors to achieve nanometer resolution). The advantage is that an ultrasonic piezo motor has zero drift after positioning and achieves a good bi-directional position repeatability.
Furthermore, the control strategy of shaping the elliptical trajectory makes it possible to follow a low but constant scanning speed of the slider. The term ‘ultrasonic’ means that the frequency of oscillation lies outside of the audible frequency range for humans. This explains why these motors operate noiselessly, which is a strong advantage when operators work in the vicinity of the system, such as an optical microscope. Additionally, because of the high frequency of operation, one can achieve very high motion speeds of 1000 mm/s and more with an ultrasonic piezo motor. The low power consumption and thus low heat generation of this type of motor is explained by its operation at resonance, which is energetically more favorable than quasi-static operation. This is important in handheld devices and systems which need to be thermally stable, such as vacuum setups or measurement devices.
Finally, these motors can be used to cover long distances and a long lifetime, when used under proper working conditions. This is explained by the lower impact between the contact point and slider, as compared to stick-slip piezo motors.
Characteristics of the different piezo motor types
Table 1 summarizes the different characteristics of the three main piezo motor types described above.
| Ultrasonic piezo | Stick-slip piezo (inertial) | Stepper piezo (walking piezo) | |
|---|---|---|---|
| Speed | 1000 mm/s | 1 - 3 mm/s | 10 - 20 mm/s |
| Force* | 1 - 10 N | 1 - 10 N | 5 - 20 N |
| Resolution | 1 nm | 1 nm | < 1 nm |
| Travel range | 1 - 300 mm | 1 - 50 mm | 1 - 50 mm |
| Lifetime | > 1000 km | 10 - 20 km | 50 - 100 km |
| Vibrations/noise | vibrations > 20 kHz (silent) | vibrations < 5 kHz (noise + resonance risk) | vibrations < 2 kHz (noise + resonance risk) |
| Constant speed | smooth (μm/s) | saw-tooth profile | small pulses from stepping |
| Power consumption | low | moderate | moderate |
* This is the force of a single motor, of comparable size (~ 1 cm³). A common approach to increase the force is to place multiple motors in parallel. Also, a larger sized motor can achieve higher forces.
Table 1: Comparison of different piezo motor characteristics.
References
Hunstig, M. (2017). “Piezoelectric Inertia Motors – A Critical Review of History, Concepts, Design, Applications, and Perspectives”, Actuators, vol. 6, no. 1, pp. 1–35, Feb. 2017.
Santoso, A. (2014). “Simultaneous multi driving mode operation of a piezoelectric motor.” Ph.D. dissertation, KU Leuven, 2014.
Want to learn more?
Continue reading about Xeryon's proprietary ultrasonic piezo technology.



